Lukas Welsch1, Charlotte Paulus1, Anna-Maria Lauerer1, Dominik Wermers1, Johanna Kohn1, Sönke Schildt1, Daniela Hirsch2, Stefan Wagner1, Lars S. Maier1, Simon Lebek1
1Department of Internal Medicine II, University Hospital Regensburg, Germany
2Institute of Pathology, University of Regensburg, Germany
Introduction
Cardiovascular disease remains the leading cause of death worldwide, and a substantial fraction is driven by malignant arrhythmias, which highlights the need for advanced antiarrhythmic strategies. The cardiac stress-responsive kinase CaMKIIδ is a central driver of arrhythmias by increasing the sarcoplasmic reticulum (SR) Ca2+ leak and enhancing the late Na+ current. Notably, CaMKIIδ becomes hyperactive upon post-translational autophosphorylation at threonine-287. We previously generated a knock-in mouse in which CaMKIIδ is rendered phospho-resistant (T287A). Whether blocking CaMKIIδ autophosphorylation confers antiarrhythmic protection remained unclear.
Methods
We studied wildtype (WT) and CaMKIIδ-T287A mice, and isolated cardiomyocytes were stimulated with 100 nM isoprenaline (ISO). Diastolic SR Ca2+ leak was assessed by confocal laser scanning microscopy in Fluo-4 AM-loaded (10 µM) cardiomyocytes. Whole cell current clamp recordings were used to analyze action potentials. Electrically stimulated Ca2+ transients (1 Hz) were recorded by epifluorescence microscopy in Fura-2 AM-loaded (5 µM) cardiomyocytes. In vivo arrhythmias were evaluated using an electrophysiological burst protocol via catheterization of the right jugular vein with an octopolar electrode catheter. The severity of arrhythmias after each burst was graded from 0 (no arrhythmias), 1 (single ventricular extrasystoles), 2 (persistent < 10s), to 3 (persistent > 10s). The arrhythmia scores from five burst stimulations were averaged to obtain an overall arrhythmia score.
Results
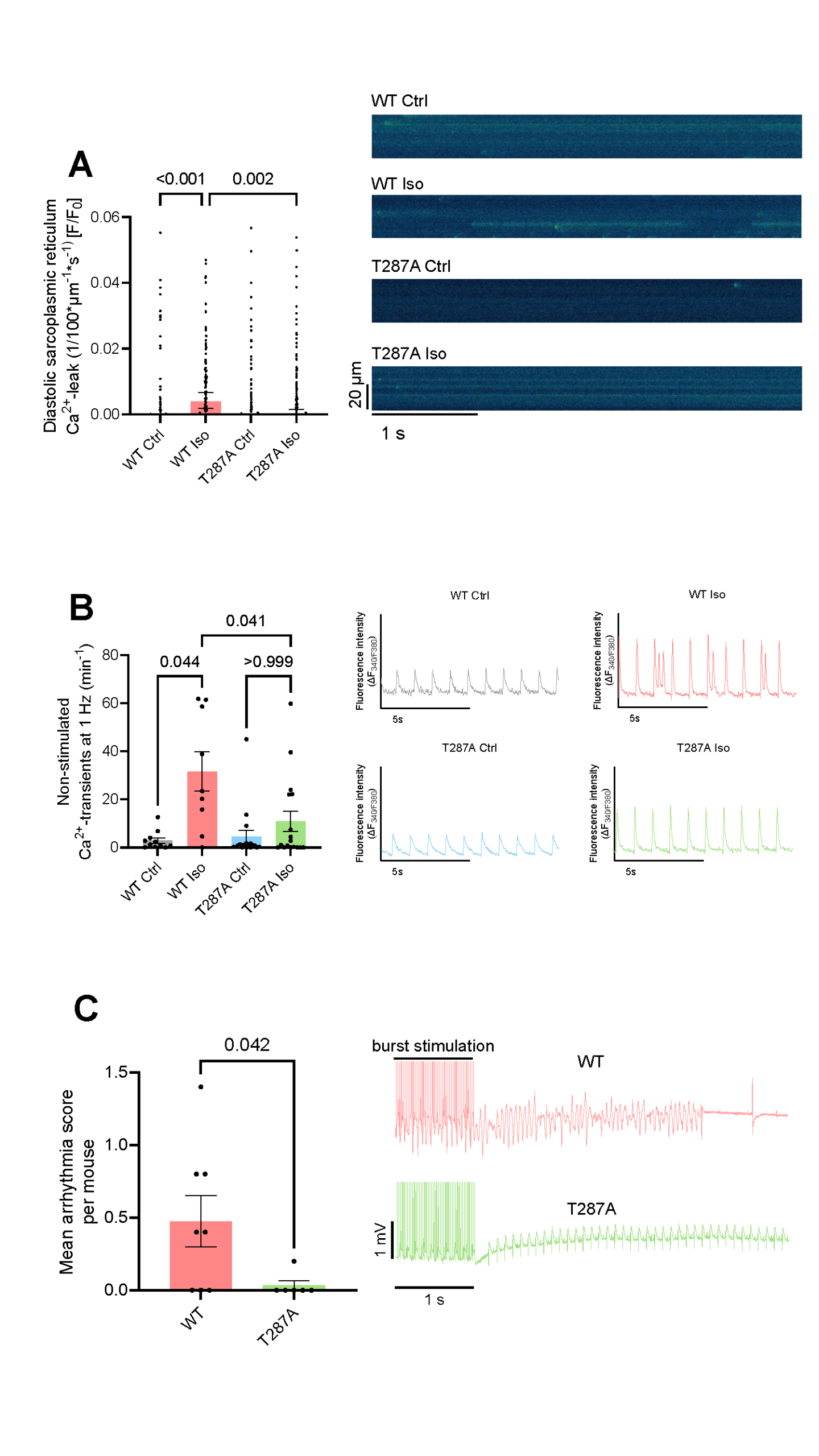
In WT cardiomyocytes, ISO caused a significant increase in Ca2+ spark frequency, which resulted in an increased diastolic SR Ca2+ leak (p<0.001, Figure 1A). In contrast, T287A cardiomyocytes were protected from these proarrhythmic alterations (Figure 1A). Patch clamp analysis of WT cardiomyocytes showed a pronounced prolongation of action potential duration at 80% repolarization from 30.1 ± 2.7 ms to 61.7 ± 4.3 ms (p<0.001) after ISO, whereas T287A cardiomyocytes were protected from this effect (44.4 ± 5.0 ms upon ISO, p=0.006 vs. WT upon ISO).
ISO exposure increased Ca2+ transient amplitude in both WT (from [ΔF340/F380] 0.17 ± 0.02 to 0.34 ± 0.04, p=0.002) and T287A cardiomyocytes (from 0.19 ± 0.02 to 0.35 ± 0.03, p<0.001). In WT cardiomyocytes, ISO led to an 11-fold increase in proarrhythmic non-stimulated Ca2+ transients (p=0.044, Figure 1B). Notably, T287A cardiomyocytes were protected from this effect upon ISO and showed a significantly lower frequency of proarrhythmic non-stimulated Ca2+ transients compared with WT cardiomyocytes (p=0.041, Figure 1B).
To increase the translational relevance, we next assessed arrhythmias in vivo in WT and T287A mice. In line with our cellular findings, T287A mice showed a substantial lower arrhythmia severity compared with WT mice (p=0.042).
ConclusionCardiomyocytes in which CaMKIIδ is phospho-resistant (T287A) are protected from an increased diastolic SR Ca2+ leak, prolongation of action potential duration, and proarrhythmic non-stimulated Ca2+ transients under acute β-adrenergic stress. These cellular mechanisms correspond to a protection against arrhythmias in vivoin T287A mice. These data support the translational potential of targeting CaMKIIδ autophosphorylation as an antiarrhythmic strategy, and ongoing work aims to ablate CaMKIIδ autophosphorylation postnatally in models of cardiovascular disease.